
Progetto di ricerca scientifica sostenuto nel 2021 da IL CUORE GRANDE DI FLAVIO – ODV
Anche quest’anno, come da Statuto, l’associazione IL CUORE GRANDE DI FLAVIO ha donato all’Ospedale Pediatrico Bambin Gesù di Roma una somma pari a 50 mila euro a sostegno della ricerca scientifica oncologica pediatrica.
Qui di seguito il progetto di ricerca finanziato attraverso la somma percepita dall’Agenzia delle Entrate con il 5×1000, annualità 2020 (24.390,49 euro) e la generosità dei sostenitori dell’associazione che non smetteremo mai di ringraziare.
Per il futuro speriamo di poter contare su entrate da 5×1000 ancora maggiori, ricordando che per donare il 5×1000 nella dichiarazione dei redditi occorre semplicemente indicare il codice fiscale 97838510580 nell’apposita casella.
Studio del micro-ambiente tumorale e della variabilità genetica delle cellule neoplasiche dopo chemioterapia nei sarcomi delle parti molli pediatrici.
Premessa
I sarcomi delle parti molli rappresentano l’8-10 % dei tumori solidi dell’età pediatrica ed includono numerosi istotipi. Tradizionalmente l’approccio terapeutico è stato basato su tipo istologico (rabdomiosarcomi e sarcomi non rabdo),stadio di malattia ed età.L’avvento di tecniche di caratterizzazione molecolare sta profondamente rivoluzionando le modalità classificative tradizionali contribuendo ad una ridefinizione dei sarcomi pediatrici sulla base non della semplice istologia, ma con l’integrazione di caratteristiche molecolari.In particolare i rabdomiosarcomi precedentemente classificati come alveolari, sulla base delle caratteristiche delle cellule ed il pattern di aggregazione, ora vengono definiti sulla base della presenza di una fusione genica PAX3/PAX7-FOXO1 (o fusioni più rare ) alveolari, o , se privi della fusione, vengono considerati rabdomiosarcomi “fusion negative” e considerati nello spettro morfologico degli embrionali ai fini terapeutici.Altrettanto per i sarcomi non rabdo, molti istotipi sono definiti dalla presenza di un’alterazione genica ricorrente. Inoltre alcune alterazioni geniche costituiscono dei bersagli di specifici trattamenti. Accanto ai protocolli terapeutici standard stanno emergendo nuove strategie di trattamento, miranti a colpire i meccanismi molecolari che mantengono la replicazione delle cellule tumorali o le proteggono dall’attacco del sistema immunitario o le rendono più resistenti alle terapie. Queste rappresentano spesso lo strumento per il salvataggio di pazienti in recidiva tumorale o progressione di malattia. Uno degli elementi chiave per la comprensione dei meccanismi di progressione e resistenza alla terapia nei sarcomi pediatrici e per la definizione di terapie personalizzate è la comprensione delle caratteristiche biologiche di tutto il tumore. Studi recenti evidenziano che le neoplasie maligne non sono sempre omogenee, ma possono essere costituite da multiple componenti cellulari con alterazioni geniche acquisite, che determinano la formazione di “cloni” con caratteristiche di resistenza alla terapia o maggiore aggressività. Nell’ambito dei sarcomi pediatrici l’eterogeneità intratumorale e la selezione di cloni nelle diverse fasi della malattia è stata finora poco esplorata. Il nostro studio si propone di analizzare le caratteristiche molecolari di una serie di sarcomi pediatrici studiando specificamente le caratteristiche molecolari all’esordio, nel residuo tumorale post-terapia e nelle recidive.
Gli obiettivi dell’analisi sono i seguenti:
-identificare aspetti molecolari caratteristici e potenzialmente target di specifiche terapie
-definire l’evoluzione molecolare del tumore dopo terapia, nell’eventuale residuo tumorale alla chirurgia e/o nella recidiva
-esplorare aree diverse del tumore per definire l’eterogeneita intratumorale e definire le caratteristiche degli eventuali meccanismi di resistenza alla terapia
Metodi
Lo studio prospettico si focalizzerà su sarcomi pediatrici rabdo e non rabdo diagnosticati presso OPBG o in altre sedi ed inviati per second opinion, previo ottenimento del consenso informato, nell anno 2022. Retrospettivamente saranno selezionati i casi di sarcoma con materiale biologico disponibile (blocchetti in paraffina o congelato) del tumore all’esordio, della chirurgia post-terapia e/o della recidiva.Per tutti i casi verrà effettuata una caratterizzazione molecolare, quando non eseguita già alla diagnosi mediante sequenziamento dell’RNA (RNAseq) e uno studio del profilo di metilazione (Figura 1). Tale analisi verrà eseguita su:
- Tumore all’esordio
- Residuo tumorale post-terapia
- Recidiva tumorale
Si procederà a rivalutare i preparati istologici in modo da definire aree morfologicamente diverse e significative per effettuare le analisi su tutte e valutare le caratteristiche molecolari e di metilazione. Questo consentirà quindi di definire aspetti di eterogeneità intratumorale. I risultati di RNAseq e di metilazione verrano integrati attraverso un’analisi bioinformatica che consentirà di definire: -fusioni geniche ed altre alterazioni molecolari -profili di espressione genica-meccanismi di metilazione coinvolti nell’espressione genicaLa comparazione dei dati nei campioni alla diagnosi, dopo terapia e nelle recidive definirà l’evoluzione della lesione, le caratteristiche dei diversi cloni (se presenti) e i meccanismi potenzialmente attaccabili con terapie mirate.

 Figura 1– Lo studio dell’epigenoma di una cellula viene effettuato mediante analisi della metilazione dell’intero genoma cellulare (Genome-Wide DNA Methylation Analysis). L’analisi del metiloma Genome-wide DNA è relativamente semplice da portare avanti, grazie a una reazione chimica, la conversione con bisulfito di sodio (A), che permette di discriminare le citosine metilate e non metilata nelle isole CpG dell’intero genoma e successivamente sfruttando la tecnologia array (B). Lo studio dei profili di metilazione sarà effettuato presso i laboratori di San Paolo dove è già disponibile lo strumento iScan (Illumina) (C), mediante Illumina Infinium HumanMethylation850K BeadChip array (D) che contenengono circa ~850.000 sonde per ogni silicon-based array.
Figura 1– Lo studio dell’epigenoma di una cellula viene effettuato mediante analisi della metilazione dell’intero genoma cellulare (Genome-Wide DNA Methylation Analysis). L’analisi del metiloma Genome-wide DNA è relativamente semplice da portare avanti, grazie a una reazione chimica, la conversione con bisulfito di sodio (A), che permette di discriminare le citosine metilate e non metilata nelle isole CpG dell’intero genoma e successivamente sfruttando la tecnologia array (B). Lo studio dei profili di metilazione sarà effettuato presso i laboratori di San Paolo dove è già disponibile lo strumento iScan (Illumina) (C), mediante Illumina Infinium HumanMethylation850K BeadChip array (D) che contenengono circa ~850.000 sonde per ogni silicon-based array.
Fattibilità
Nel 2020-2021 sono stati caratterizzati mediante RNAseq e metilazione (Figura 2) circa 80 sarcomi alla diagnosi. Questo ha consentito di definire caratteristiche molecolari all’esordio ed identificare alterazioni molecolari ricorrenti non ancor descritte in circa 2% di esse (alcune già pubblicate). Saranno selezionati i casi con disponibilità di campione post-operatorio e/o recidiva con completamento delle analisi come descritto nei metodi.
Per lo studio prospettico, il progetto descritto verrà eseguito come WP5 (working package 5) in uno studio multicentrico europeo (Mykids) sui sarcomi non-rabdo e questo consentirà il reclutamento di un numero di pazienti ampio.
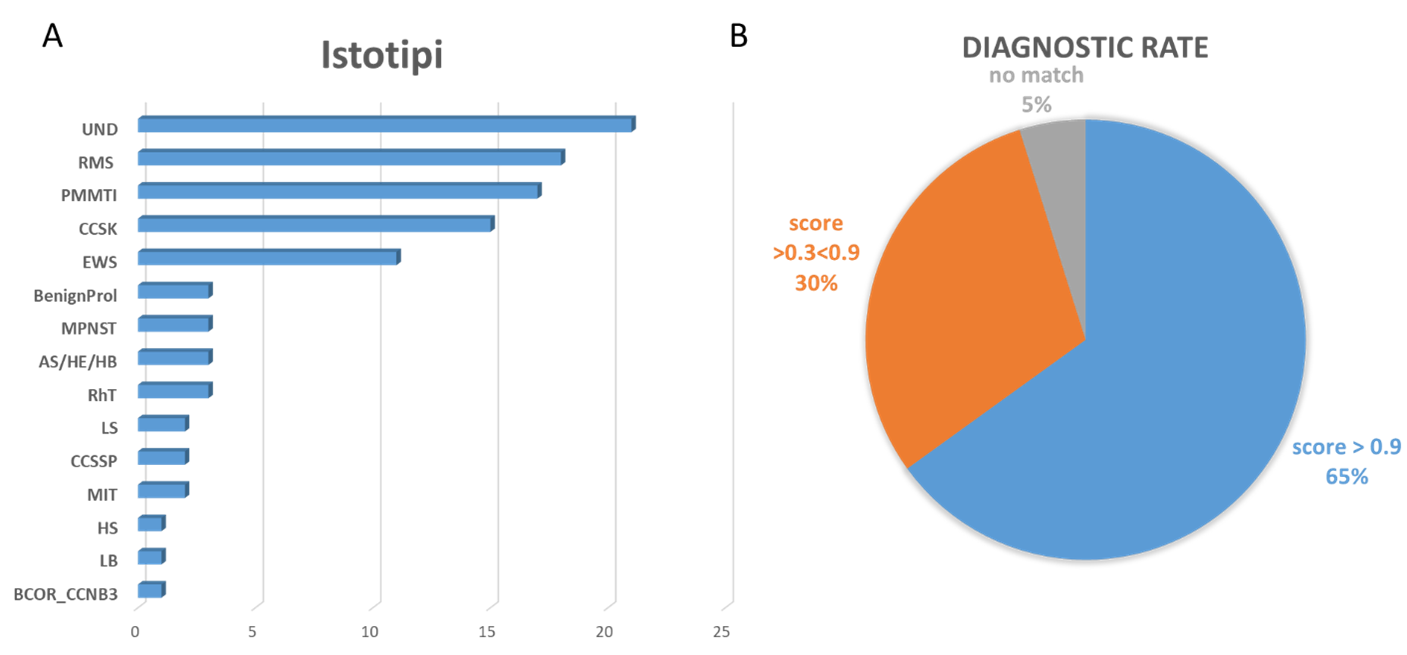
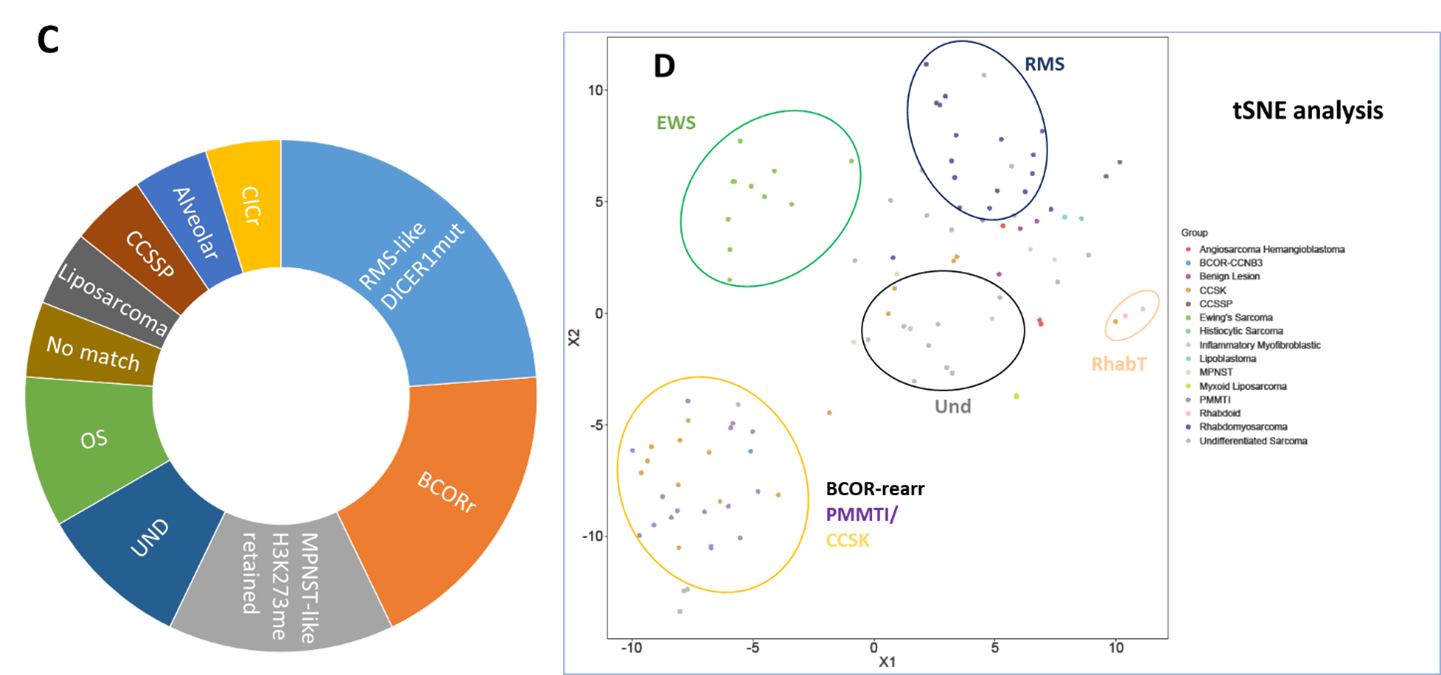
Figura 2. Gli istotipi analizzati sono mostrati nel pannello A mentre il tasso diagnostico ottenuto confrontando i nostri risultati con quelli del classificatore di sarcoma di Heidelberg sono mostrati nel pannello B. I nostri campioni sono stati attribuiti a una classe di metilazione identificata con un punteggio ottimale nel 65% dei casi, nel 30% il classificatore ha orientato la diagnosi in una classe di metilazione con un punteggio maggiore di 0,3, mentre solo il 5% dei casi è risultato inclassificabile. C) Il profilo di metilazione del DNA ha permesso un orientamento diagnostico e o un perfezionamento diagnostico, principalmente per tumori indifferenziati. Con l’eccezione di un caso, tutti i sarcomi indifferenziati analizzati sono stati inclusi in classi di metilazione specifiche e la diagnosi è stata successivamente confermata molecolarmente mediante altri test. D) Abbiamo inoltre analizzato i nostri campioni utilizzando l’analisi di clustering con l’algoritmo t-SNE. Abbiamo identificato chiari clusters rappresentati da sarcomi EW canonici, tumori riarrangiati BCOR tra cui sia tumore mesenchimale mixoide primitivo dell’infanzia (PMMTI)/ e sarcoma cellulare chiaro del rene, un cluster che include la maggior parte dei sarcomi-rabdo, uno per il tumore Rhabdoidi, e un cluster che include alcuni tumori indifferenziati.
Risultati Attesi
Lo scopo dello studio è di reclutare un numero di pazienti adeguato per ciascun istotipo tumorale (almeno 10) nel complesso ci si aspetta di studiare 6 istotipi (Rabdo e non-rabdo), con un numero di RNAseq e profili di metilazione atteso per ciascun caso da 2 a 6 a seconda della disponibilità del materiale. Ci si attende di poter ricostruire l’evoluzione molecolare della neoplasia e definire i pathway molecolari che alla diagnosi possono essere indicativi di aggressività e pertanto richiedono adeguato approccio terapeutico. (Figura 3).
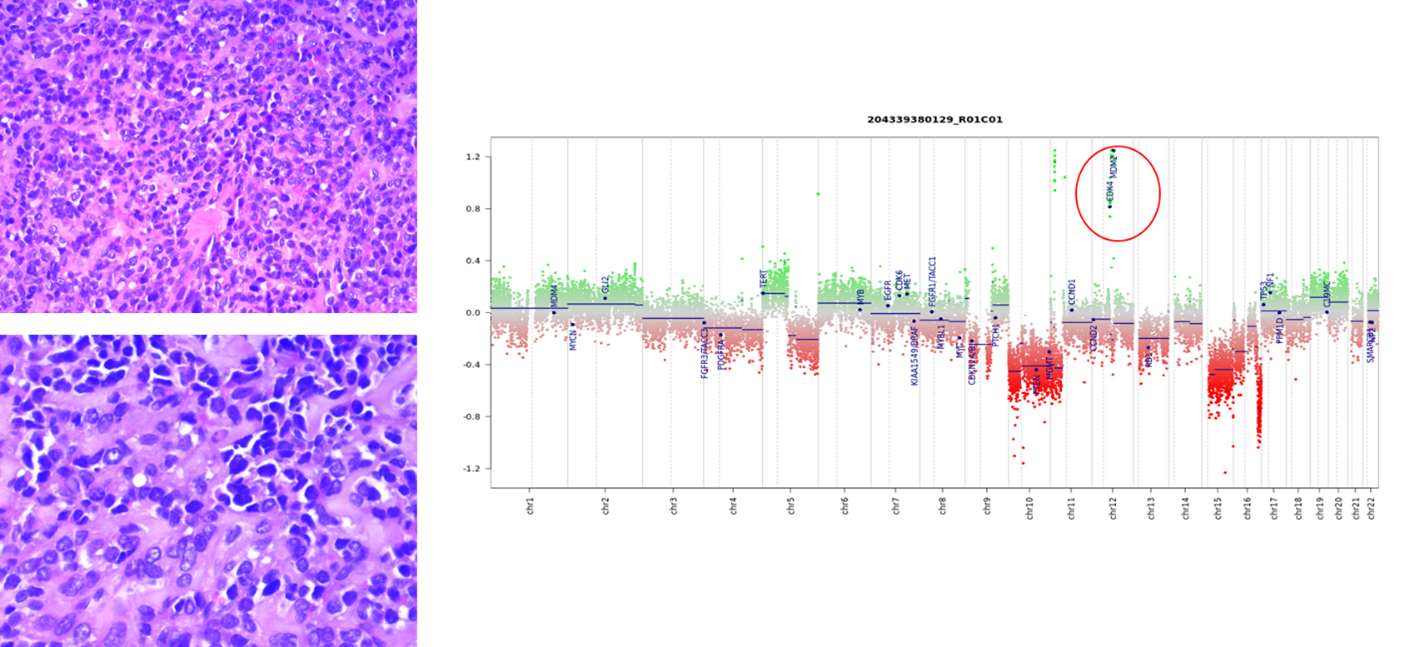
Figura 3. Dall’analisi degli array di metilazione del DNA è possibile dedurre informazioni anche sulla variazione del numero di copia del DNA, un parametro importante per stabilire l’aggressività del tumore.
La figura mostra un esempio di amplificazione genica (CDK4 e MDM2, nei cerchi rossi) in un caso di rabdomiosarcoma sclerosante con mutazione MYOD1.
